Setting Up New Online Account and Contact Guideline
At anytime, you can register for a new online account at https://order.apicalscientific.com/ and directly placing an order.
For overseas customers, our authorised local distributor(s) will be listed in your respective country during new account registration to ease billing in your local currency and expedite order submission.
You can email or contact us as below:
| Malaysia | custcare@apicalscientific.com | Call or WhatsApp +603-8943-3252 |
| Other Countries | Contact our respective sales representative in your region | |
Our online ordering system enables direct order of 1st BASE Services such as Standard Sequencing Services, Sequencing+ PLUS Services, Direct Colony Sequencing, Primer Walking, DNA Barcoding Services and Avian DNA Testing.
The setup of online order for life science products and reagents will be available soon.
We had transferred the primary email address of active customers as the new login ID into this new online ordering system. If you do not receive an email notification and step-by-step guideline to confirm the new registration, please contact us using LiveChat or WhatsApp during our business hours for direct assistance.
For customers who ship or courier us the orders directly, please indicate your airway bill number or courier tracking number in your online order directly or drop us your email by indicating the respective online order ID to ensure speedy processing of your orders.
For overseas customers who pay the orders through our authorised distributor(s), please contact the respective distributor by indicating your online order ID to process the orders according to the local arrangement of your country.
Kindly refer our order submission guideline for details.
We have prepared a series of tutorial videos to assist you in your ordering process, please click on the link below to be directed to the playlist.
FAQ for Sanger DNA Sequencing Services
We offer trials for new customers to try out our Sanger DNA sequencing services. Please contact our sales representative in your region to find out more.
Direct customers will be able to view the price upon ordering on our Online Ordering system. If you are placing an order with us for the first time, the displayed price online might not be updated. Please do not worry as you can still proceed with the ordering. We will update the pricing accordingly as soon as we confirm your order.
For customers that order through our dealers or distributors, you may contact our respective sales representative in your region for pricing details.
Sequencing order collection points are set up in a few locations in Malaysia. Please contact our respective sales representative to find out more or if you wish to set up a new collection point.
| Malaysia | 1. Kuala Lumpur/ Klang Valley: Our dispatch will collect at your location 2. Outside of Kuala Lumpur and Klang Valley: Please ship to us via local courier service. E.g Skynet / CityLink / Poslaju etc. |
| Other Countries | Please contact the local distributor in your respective countries |
1st BASE is the leading brand of Sanger DNA sequencing services, offering high quality results at fast turnaround time. The competitive edge of 1st BASE DNA Sequencing Service includes:
-
- High quality and long read lengths (~ 1000 bases) for pure DNA template
- Fast turnaround time
- Wide range of free sequencing primers available
- 1st BASE DNA Sequencing Collection Kit available for ease of organizing DNA templates and sequencing primers before sending your order
- ISO 17025:2017 certified processes. Only QC passed and authorised results are released to our valued customer
- Online ordering system allows smooth ordering acceptance
FAQ for Sequencing Order Preparation & Submission
You can place an order with us via online:
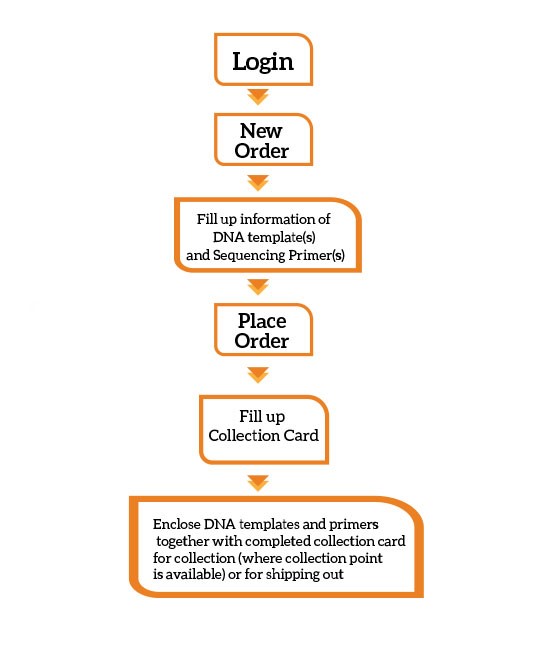
Remember to label the Online Order ID on the provided Sample Collection Card or your own packaging. Kindly refer to our quick guide for order registration and submission.
We offer comprehensive service to meet your needs! You can choose the service based on your DNA template type. If you are unsure, kindly contact us.
| Service Type | Type of DNA Template |
| Single Pass DNA Sequencing | Standard Sanger DNA sequencing service for purified DNA templates. Guaranteed read length of over 1000 bases for good quality DNA templates. |
| DNA Sequencing+ PLUS | Complete Sanger DNA sequencing solution from unpurified PCR and unpurified plasmid products. Guaranteed read length of over 1000 bases. |
| Ready-to-load Sequencing | Electrophoresis of purified or unpurified cycle sequencing products on Genetic Analyzers. |
| Difficult Template Sequencing (DTS) | (i) Optimized protocol developed for DNA template with secondary structures, high G%, high C%, high GC%, repeat regions or loop structures found in shRNA vectors. (ii) Large DNA templates like ss M13 DNA, BAC-end or genomic DNA also can be sequenced with DTS using the sequencing primer that has the proven good priming efficiency. |
| Direct Colony Sequencing | Direct sequencing of single colony from agar streaks. Guaranteed read length of over 600 bases. |
1st BASE DNA Sequencing Workflow
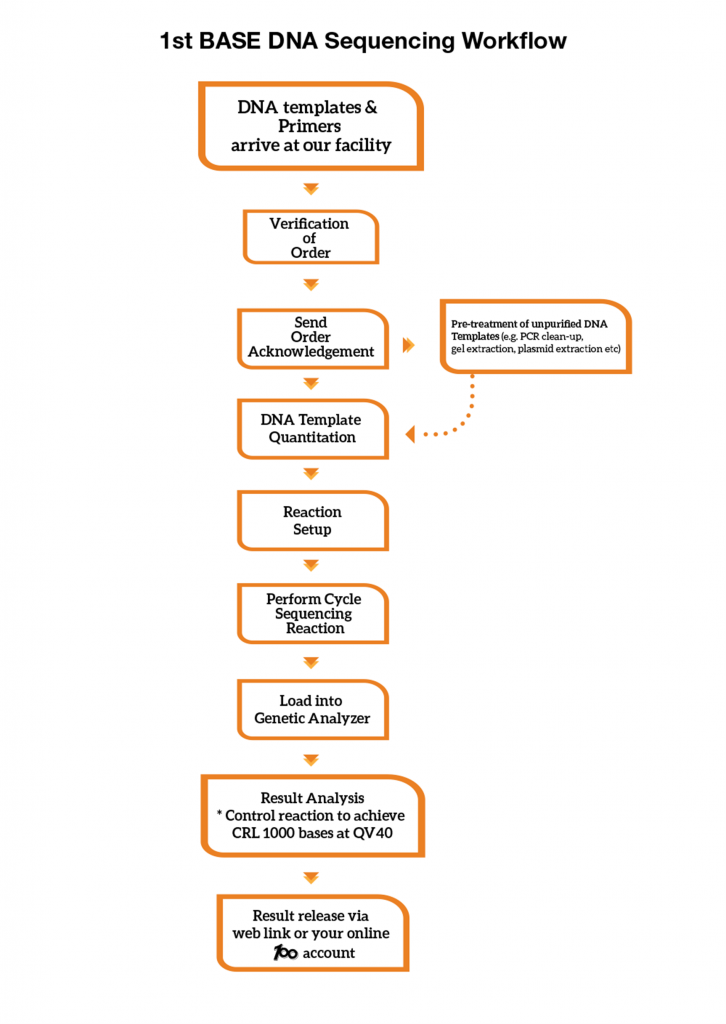
A control sequencing reaction is performed with every run in our Genetic Analyzer. Sequencing reaction controls are used to evaluate the quality of the overall reactions. Our control reaction must achieve Phred40 data and a minimum Contiguous Read Length (CRL) of 1000 bases to meet our Quality Control (QC) benchmark. We provide our control sequencing electropherogram in each of your sequencing order at no charge.
The quality of Sanger sequencing result is closely dependent on the purity and optimum amount of DNA template.
For plasmid DNA:
Most commercial DNA clean-up kits will give DNA of sufficient quality for Sanger sequencing. The most common problem arises from excess salts which usually occur if DNA was eluted in a high salt buffer or diluted/suspended in TE buffers. For Sanger DNA sequencing, we recommend keeping the plasmid DNA in 10mM Tris-HCI pH8.0 buffer or deionized water. TE buffer also can be used to keep the plasmid DNA; but you must pay extra attention for low copy plasmid, where EDTA from the TE buffer may weaken the sequencing signal. Low EDTA TE buffer or TE Buffer with reduced EDTA will be a better option.
Alternatively, we provide plasmid extraction service as an add-on service.
For PCR products:
PCR clean-up is required to ensure that the DNA is free of contaminants, excess PCR primers and dNTPs. Unpurified PCR products are not suitable for direct sequencing as the excess PCR primers will interfere the sequencing signals, which the DNA template supposed to be sequenced by the assigned single sequencing primer for each sequencing reaction. It is also highly recommended that an agarose gel is run to verify that the PCR products is single band with correct target size. Purification can be done using any commercially available kits for gel extraction or PCR clean-up. Alternatively, we provide PCR clean-up and gel extraction service as an add-on service.
Kindly refer our order preparation guideline for details.
If the residues of the PCR primers were depleted after the PCR, the noise background generated during the cycle sequencing may not affect the major base calls. However, the presence of the other contaminants (e.g. dNTPs, salts and etc.) could still compromise the quality of the DNA sequencing result to some extents.
If your unpurified PCR products is single DNA band product on agarose gel, you may order DNA Sequencing + PLUS directly. We will purify the PCR products and enable the remaining DNA to be sequenced immediately in the same sequencing order.
If you wish to verify the unpurified PCR products with only 1 sequencing primer and want the purified PCR products to be sequenced using other sequencing primer in the next new sequencing order, you must order Single Pass Sequencing with add-on PCR clean-up purification service in the first place; so that there will be sufficient purified PCR products to be sequenced in your next new order. Do note that we will only keep the remaining DNA template for 1 week after the results was released. Thus, it will be much straight forward to have all your desired sequencing primers used in the first sequencing order to minimize delays and avoid disappointment.
It is not necessary to lyophilize or dry your DNA templates before Sanger DNA sequencing services. Please submit your DNA templates in deionized water or low EDTA buffer at room temperature. DNA is stable in water at room temperature for a few days.
The quality of DNA template is the most common issue in problematic Sanger DNA sequencing reads. The amount of DNA templates used in sequencing reaction can affect the quality of data. Too little DNA template will generate low signal strength while too much DNA templates will generate a ski-slope effect which causes the signal to weaken gradually.
It is essential to accurately quantitate DNA templates prior to sequencing. However, accurate quantification of dsDNA is not easy.
It’s informative to run your DNA template on an agarose gel, using some standard of known concentration and estimate the relative fluorescent intensity following gel staining. DNA ladders/markers can be used as a reference standard since it comes with a known amount of DNA (ng) for each DNA band.
Kindly refer our guidelines to read the agarose gel electrophoresis for details.
The regular gel photo shall display different DNA intensity for its known DNA Ladder, so that we can use it to estimate the DNA amount of your purified PCR products that loaded next to the DNA Ladder.
By using the example of gel photos below, when we compare the DNA intensity of the purified PCR Products with its DNA Ladder: the known DNA size of 600bp (from DNA Ladder) near to the target PCR products is 12ng and it looks brighter, we can thus confidently say that the purified PCR Products has < 12ng of DNA. If we load only 1µL of purified PCR Products in this agarose gel, we can then estimate the DNA concentration of the purified PCR Products is < 12ng/ µL, which you can take any number between 5ng/µL to 9ng/µL, but not any figure near 12ng/µL.

If the agarose gel is over-exposed under the UV light during the gel photo is taken by any gel documentation system, the displayed intensity of the DNA ladder will not be able to guide us to estimate the DNA amount confidently. Although the DNA intensity of the purified PCR products is higher in this gel photo, but the actual DNA concentration is only 5ng/µL to 9ng/µL under regular gel photo.
Generally, there are 2 commonly used methods to quantitate DNA templates. But neither method will show the presence of contaminating salts.
-
- Agarose gel electrophoresis – Purified DNA should run as a single band on an agarose gel (Uncut plasmid DNA can be visualized as 3 bands: supercoiled, nicked, and liner). Contaminating DNA or RNA will show up in the gel.
- UV Spectrophotometry - The A260/A280 ratio should be between 1.7 and 1.9. Smaller ratios usually indicate the presence of contaminating proteins or phenol. The NanoDrop ND-1000 spectrophotometer takes 1 to 2 µL of sample and can detect concentrations from 2 to 3700 ng/µl without dilutions.
Traditional absorbance invariably overestimates DNA. In a finding, Kapp et al (2014) encounter a five-fold difference between absorbance-based method and fluorescent-based method, which could theoretically result in using lower amounts of starting DNA for the downstream sequencing step and cause unsatisfactory results. Discrepancies between the two methods were also reported by Deben et al, O'Neill et al and Simbolo et al (2013). The measurements at 260nm (A260) fluctuate with contaminants and buffer component, mainly because miniprep kits on the market often inevitably introduce some RNA, chromosomal DNA, and other fluorescent cellular material during the purification process that will absorb UV light. These other chemicals won’t necessarily inhibit the sequencing reaction, but they will contribute to the A260 reading. As a result, relying on the spectrophotometer reading alone will cause you to add less DNA template than you think.
Fluorescence assays on the other hand are less prone to interference. Fluorescence-based quantitation is more sensitive and more specific for the nucleic acid of interest. In the technical note from Invitrogen, comparison between the UV absorbance method and the fluorescent method was compared. Nanodrop method was found to shown fluctuation particularly at lower concentration range. Whereas, fluorescence-based quantitation generate accurate and precise result across lower concentration range.
Our laboratories adopt fluorescence-based measurement which might explain the difference in the measured DNA concentration. We take every step to ensure that the quantification process is consistent and accurate for downstream processes. Our laboratory apparatus is calibrated and maintained regularly to ensure they are working within their specified limits.
References:
Kapp JR, Diss T, Spicer J, et al Variation in pre-PCR processing of FFPE samples leads to discrepancies in BRAF and EGFR mutation detection: a diagnostic RING trial Journal of Clinical Pathology Published Online First: 27 November 2014. doi: 10.1136/jclinpath-2014-202644
Simbolo M, Gottardi M, Corbo V, et al. DNA qualification workflow for next generation sequencing of histopathological samples. PLoS ONE 2013;8:e62692
Invitrogen™ Technical Note. Comparison of fluorescence-based quantitation with UV absorbance measurements: Qubit™ Quantitation Platform vs. spectrophotometer
We do not recommend sending DNA template of insufficient amount. If you request us to process such DNA template to sequencing, we will try to deliver our best effort guarantee to maximize the success of your sequencing order. Although a specialized protocol will be deployed to maximize the amplification (and there are just so much we can do), we are still unable to guarantee optimal DNA sequencing result.
Ideally DNA templates should be 100bp and above. With our optimized running protocol and module, we can sequence DNA templates as small as 90-100bp, but this will be subjected to the DNA template quality and the priming efficiency of its sequencing primer.
For DNA templates less than 90bp, we recommend the sequencing to be done using Next-Generation Sequencing.
We will only keep DNA templates and sequencing primers for 1 week upon receiving them. Please indicate in the order form if you would like us to keep your DNA templates or sequencing primers for further sequencing reactions. A nominal fee will apply for storing your DNA templates or sequencing primers in our laboratories.
FAQ for Sequencing Results
Upon the receipt of your sequencing order to our DNA sequencing facility, an order confirmation will be sent to you through email. We typically deliver the sequencing result to you within 48 working hours upon the receipt of your DNA sequencing order with complete ordering information. For ready-to-load reactions, results are typically ready within 24 working hours.
Kindly refer our list of DNA sequencing services for details.
Results will be downloaded through Online Ordering account. The compressed file in .zip format (for Windows), contains the complete sequencing results with (i) electropherogram(s) in .ab1 file and (ii) fasta sequence in .seq file.
We understand that the confidentiality of your result data is critical. To ensure a high level of security towards the result uploaded, for customers who yet to register an online account with us, we would provide result in email with the weblink that is password protected. The password will only reveal to the email recipient.
To view the electropherogram that saved as .ab1 file, we recommend freeware from Applied Biosystems® Sequence Scanner 2.0 for Windows 10 or above, which enable automatic Quality Control Report at a glance. Anyhow, this file can also be viewed by other freeware like Chromas, FinchTV, and etc.
To view the fasta sequence that saved as .seq file, Notepad or Text can be used.
The results stored in the online account has expiry of 12 months. For customers who do not have the online account, the web link provided in the email to download the results has a validity of 3 months.
Please contact us through LiveChat or email by indicating your online sequencing order ID and the registered email address of recipient for verification, so another access code can be provided to you.
Please contact us through LiveChat or email by indicating your online sequencing order ID, so that we could assist you directly.
FAQ for DNA Sequencing Primers
Ideally, sequencing primer should be designed 100 bases upstream of your sequence of interest. Below are some other consideration when doing your primer design for sequencing:
- Length of primer to be between 18-25 bases
- GC content to be between 40-60%
- Melting temperature at around 55-60°C
- Check for significant hairpins (>3bp) and homodimerization
- Choose your sequencing primer desalted from Integrated DNA Technologies (IDT) or HPLC grade from other manufacturers
Many commercial oligo providers offer online design tools which you can use. We recommend IDT’s OligoDesigner.
DNA Sequencing Results Interpretation
& Troubleshooting
For .seq file, it is the nucleotide fasta sequences extracted from electropherogram, and can view by Notepad or Text.
For .ab1 file, it is the electropherogram or sequencing trace file that requires specific software to view. We highly recommend using Applied Biosystems® Sequence Scanner 2.0 software to view any Sanger electropherogram or sequencing trace file. The weblink to download this freeware is provided with each release of 1st BASE DNA Sequencing Order. There is a minimum system requirement to install this software completely. The installation window will check your computer system during installation. Alternatively, the electropherogram can also be viewed using Chromas, FinchTV, DNA Baser and etc.
It is important to identify what calls as a good sequencing trace. Our sequencing results show you the confidence of each base called by using KB Basecaller software. The confidence level is showed as quality value (QV) for each base.
Trace view for good sequence using Sequence Scanner software:
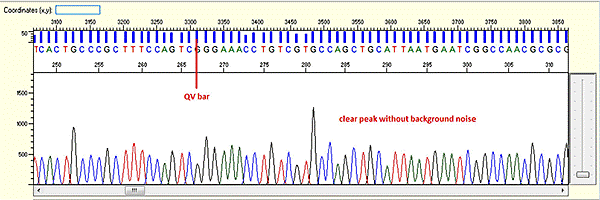
A good sequencing trace data will contain: -
-
- Continuous long stretch or un-interrupted good quality value (QV) of the basecall. The QV will be displayed as rectangle bars at the upper part of the electropherogram.There are 3 categories of QV bars: -
- Blue: high quality with >= QV 20
- Yellow: medium quality with QV 15 to 19
- Red: low quality with QV < 15
- Well defined peaks with almost no or very minimum of background signal.
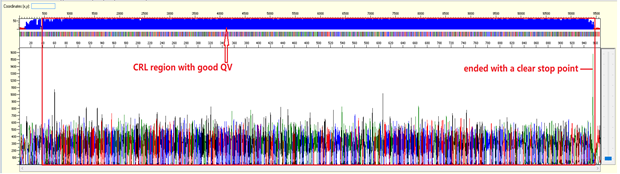
For sequencing trace of plasmid DNA or long PCR product (>1000bp), we will not be able to see the end of the trace (or we called as stop point) as the template size is larger than what the electrophoresis can separate. There should be a continuous read length (CRL) with good QV until 850 bases, then the resolution of the electrophoresis will gradually loss and there will be more low quality QVs.
- Continuous long stretch or un-interrupted good quality value (QV) of the basecall. The QV will be displayed as rectangle bars at the upper part of the electropherogram.There are 3 categories of QV bars: -
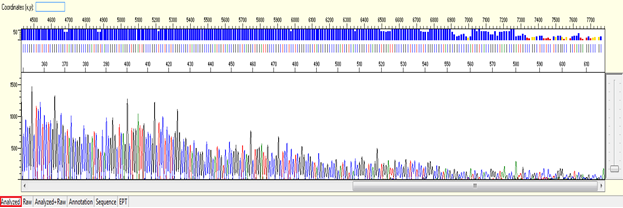
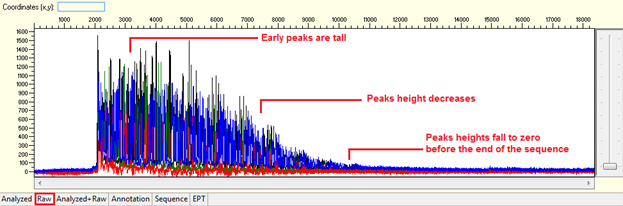
Pattern: Ski slope configuration of the trace.
The sequence starts with high quality peaks but become messy in the downstream. The raw data will show a good signal intensity at the beginning of the sequence and decreases when the sequence continues.
Trace view for early signal loss sequence using Sequence Scanner software:
1. Causes:
-
- Improper amount of DNA template.
- Present of contaminants in the DNA template / sequencing primer.
- Difficult region within the DNA template.
- Degrading DNA template.
Solutions:
-
- We 100% quantify each DNA template before cycle sequencing. We will thus be able to calculate the optimum volume required for each DNA template. Thus, providing us the correct or approximate size (less than 100 bases different from the actual size) of DNA template will prevent too much/ too little DNA template used in the cycle sequencing reaction. Please refer the guideline provided in our Order Preparation and Submission.
- Any commercial column purification kit will be able to remove contaminants effectively. A good advice for researcher that purifying bulk samples, we highly recommend processing the purification in several batches carefully. For purification kit using high salt as binding buffer, a skilful laboratory practitioner will prevent salts from contaminating the final elution. Alternatively, we also offer Sample Preparation and Quantification Services before sequencing.
Pattern: The signal is good before, and it suddenly terminates or drops without continued basecalling.
Trace view for abrupt signal loss sequence using Sequence Scanner software:
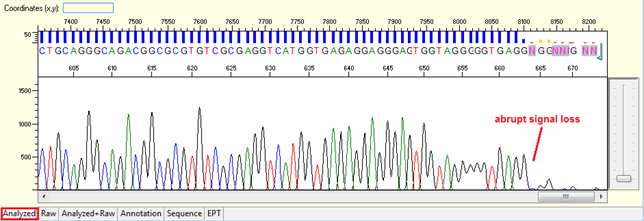
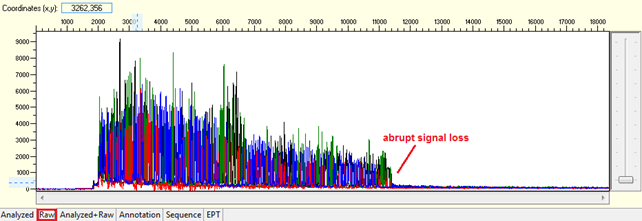
Causes:
-
- Difficult region found in the sequence, which stops the polymerase reaction.
- Formation of DNA secondary structure.
Solutions:
-
- Sequence from the opposite strand.
- Order our Difficult Template Sequencing Service, which uses an alternative cycle protocol to sequence the difficult regions.
- Difficult region and DNA secondary structure from PCR product template tend to fail to be sequenced; cloning will be an option to get stable and thus longer read.
Alternatively, we provide DNA cloning under our Molecular Biology Services before cycle sequencing. Please enquire.
Pattern for noisy data with weak signal: Undefined peaks, no continuous read length (CRL), and majority having low quality QV bars.
Trace view for noisy data with weak signal sequence using Sequence Scanner software:
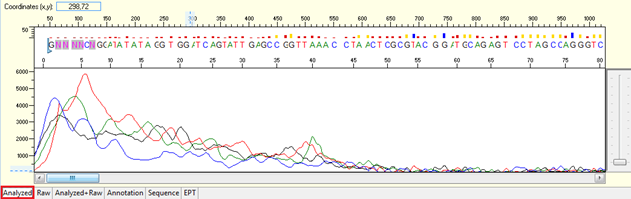
Pattern for no usable signal: Data showed only 5Ns. The sequence trace file may not be able to open by using certain software.
Trace view for no usable signal sequence using Sequence Scanner software:
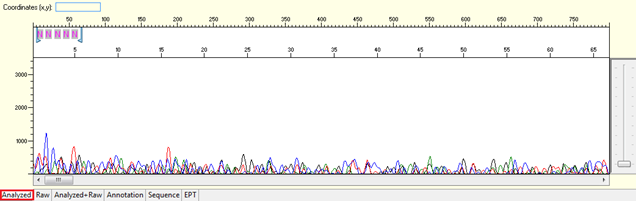
Causes:
-
- Too little DNA template or sequencing primer in the reaction.
- Inhibitory contaminant present in template.
Present of contaminant (e.g salt, ethanol, phenol, RNA, etc.) will inhibit the binding of sequencing primer or polymerase to the DNA template to start the reaction. - Degraded DNA template or sequencing primer.
- No priming site/ weak priming efficiency.
Solutions:
-
- Quantify your DNA template through gel electrophoresis is highly recommended. For short DNA templates, as long as there is a visible DNA band when 1µL of your purified DNA loaded in agarose gel, it will be sufficient to be sequenced. However, large DNA templates will require more DNA amount to get the optimum reads. Please refer the guideline provided in our Order Preparation and Submission.
- Checking the purity of the dsDNA at ODA260/280 ratio with spectrophotometer e.g Nanodrop is recommended. A pure dsDNA should have ODA260/280 ratio between 1.8 to 2.0. In our opinion, if you are using a commercial kit, and they are not expired, the quality of the purified sample should be working well for sequencing typically. The contamination that leads to weak/ no usable signal usually is the serious degradation of DNA templates.
- Contamination with DNase tend to cause different degree of degradation. Elution with TE buffer can help to protect your DNA from degradation, but EDTA in the TE buffer is one of the inhibitors for sequencing. To avoid inhibitory effect of EDTA in the sequencing reaction, you must make sure the DNA amount that present in the sample is higher/ fall under our sample requirement.
- If you are sequencing a purified plasmid DNA, we highly recommend using the vector’s sequencing primer for your first reaction to avoid disappointment. For purified PCR products, most of the researchers will use one of their PCR primers as sequencing primer. If the design of PCR primers also fulfil the requirements as a sequencing primer, it will be fine.
Pattern: Multiple overlapping peaks with strong signal from beginning. The secondary peak from the same position may be the same, lower, or higher than the major peak.
Trace view for noisy data with strong signal or high level of background noises using Sequence Scanner software:
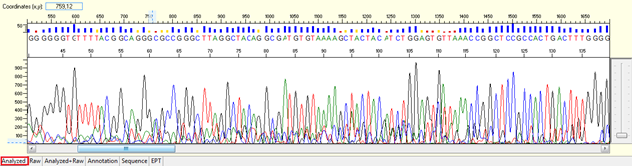
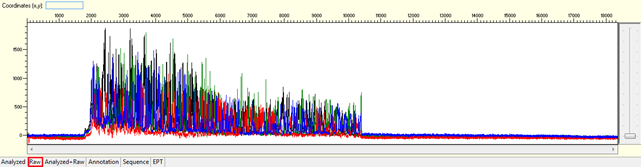
Causes:
-
- Unspecific priming site.
- Trace of excess PCR primers in the DNA template.
- Presence of more than one DNA template.
- Occasionally, we will find some purified PCR products show more than 1 one DNA band in the agarose gel photo. Most of the commercial PCR Clean-up kits will not be able to remove the contaminated double stranded DNA or primer dimer. If the sequencing primer can bind on these unspecific DNA bands or primer dimer, it will generate different cluster of extension products and being analysed together to with the major extension cycle sequencing products, which leads to mixed signal. In some cases, it is pretty easy to identify the mixed signal under this category. For example: you have an unspecific DNA band at the size of 200bp, and your target PCR product is at the size of 750bp; you will found the mixed signal will stop near the region of 180-base, following is the clean signal until the end of the sequencing trace.
- There is a mutation in your DNA template, which causing frame shift deletion in the sequencing result. 1 frame shift = 1 amino acid = 3 basepairs. If your actual target PCR products is 750bp, a frame shift deletion sample will have both 750bp and 747bp of PCR products. You will not be able to distinguish such a small different in size using any type of agarose gel electrophoresis. But the sequencing result will show different end point with 3 bases of differences. The mixed signal will start immediately after the mutation site, due to different signal from the extension products. This observation is however limited to the PCR product size. When the size of PCR products is greater than 850bp, it is very difficult to judge the different of the end point since the resolution of electrophoresis gradually loss beyond 850 bases.
- Sequencing primer with low annealing temperature.
When the Ta is lower than 48°C, we will follow your recommended Ta on the order form. The same Ta may work for your PCR, but not necessary works for sequencing. PCR is an exponential reaction that involves 2 PCR primers, but sequencing is a linear reaction that involves 1 sequencing primer.
Solutions:
-
- Redesign primer specifically for sequencing. If you are sequencing a plasmid DNA, the vector’s primer usually works very well. Alternatively, we help our customers to design and synthesis sequencing primer. Please enquire.
- Optimize the PCR protocol to have depleted primers during the end of PCR, so you can send your unpurified PCR products directly for sequencing. However, sometimes it will fail when the PCR out of control. The best safeguard is to purify your PCR products before sequencing. Alternatively, please order purification service before sequencing.
- Examine your purified DNA template on agarose gel before send for sequencing. If there is more than one DNA band observed on agarose gel, purify them with gel extraction purification rather than PCR clean-up.
- If you are bothering with the DNA template that showing mutation, the best solution is to clone the fragment and pick some colonies for sequencing. You should justify the cost of DNA cloning only if you are really interested with the mutated fragment. Otherwise, put it aside and sequence the next sample.
- If you are using one of your PCR primer as your sequencing primer, you may simply compliance the rules of sequencing primer when select the best PCR primer that designed by the primer software.
Pattern:
-
- Homopolymer: Noisy data after a long stretch of mono- (homopolymer) or di-nucleotide repeat.
- Repetitive sequence: Noisy data after a region of the template DNA that is especially high in one or two of the nucleotides.
Trace view for homopolymer (difficult template) using Sequence Scanner software:
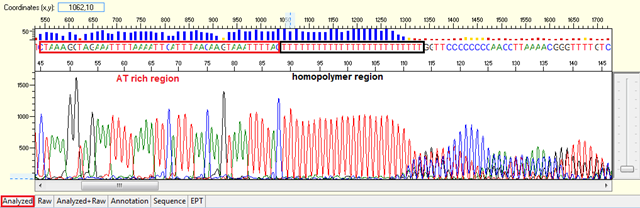
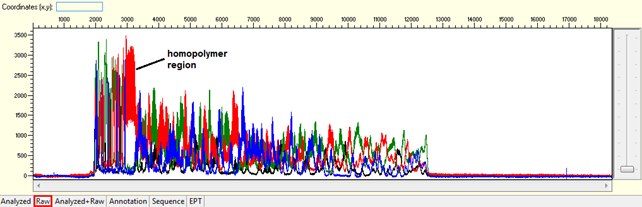
Causes:
-
- Polymerase did not pair correctly with the template.
The polymerase dissociates and re-hybridized in a different location when meet with a long stretch of mono- or di-nucleotide repeat generating various sized fragments and creating mixed signal after the region. This normally will generate a slippage pattern in the results. The condition tends to be more problematic in PCR products.
- Polymerase did not pair correctly with the template.
Solutions:
-
- For PCR products, clone the fragment into a holding vector will help to stabilize the sequencing through the same homopolymer region. It is advisable to test on a few samples firstly before use on large number of samples.
- Sequence from the opposite strand.
- Use anchored sequencing primer
Pattern: Data showing noisy peaks from the start until a specific point. The continue data showing good QVs.
Trace view for noise up to a specific point using Sequence Scanner software:
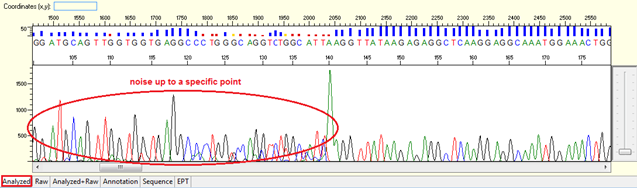
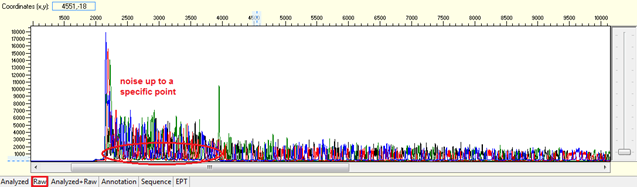
Causes:
-
- Contamination of primer dimers.
- Contamination of smaller fragment/ illegitimate products.
Soltions:
-
- The quick way is to reduce the amount of PCR primers during conduct the PCR. It works fine only if your PCR primers do not have any degenerate base(s).When you design your PCR primers, use the software to choose the best PCR primer sequence that does not perform self-dimers.
- If you able to recover this region from your opposite sequencing result, what you need to do is probably just cut off/ trim off the noise region before performing the pairwise alignment. This is workable if your DNA template is short (<850bp).
- The easy way to resolve this is to gel purify the DNA template before sequencing.
Pattern: Data is good at the beginning turns to double or more peaks after a specific position.
Trace view for noise after a specific point using Sequence Scanner software:
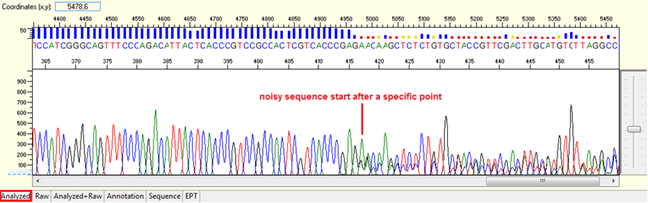
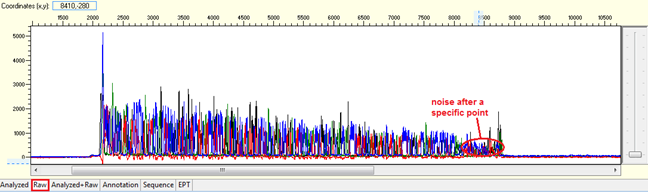
Causes:
-
- Mixed preparation of a plasmid DNA sample.
There is more than one colony is picked and extracted for sequencing. The noise is found right after the multiple cloning site (MCS). - Frameshift mutation.
Insertion or deletion of nucleotide occur in PCR product. The noise will start right after the mutation site.
- Mixed preparation of a plasmid DNA sample.
Solutions:
-
- Re-extract the plasmid DNA from the single colony to re-sequencing.
- The best solution to sequence the mutated samples is still to clone the fragments and pick few colonies for sequencing. You should justify the cost of cloning only if you are really interested with the mutated fragment. Otherwise, you should sequence another sample.
Pattern: Overlapped peaks throughout with the second peaks generally 1 base smaller and being the same base as that of the true base immediately to the right. It looks like a small “tail” before the peak of the correspondent nucleotide.
Trace view for N-1 result pattern using Sequence Scanner software:
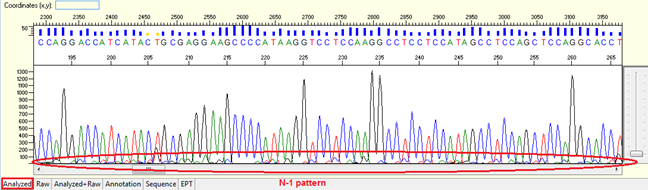
Causes:
-
- Poor synthesis quality of the sequencing primer because of low coupling efficiency.
- Mis-priming
- Degrading of sequencing primer.
Solutions:
-
- Ask replacement from your primer synthesis company. A new synthesis usually will resolve this problem immediately.
- Re-design the sequencing primer by extending the primer length will help to increase the priming specificity. We can certainly help you in the sequencing primer design and synthesis. Please enquire.
Pattern: The sequence contain dye blob may or may not affect the basecalling.
Trace view for dye blob result using Sequence Scanner software:
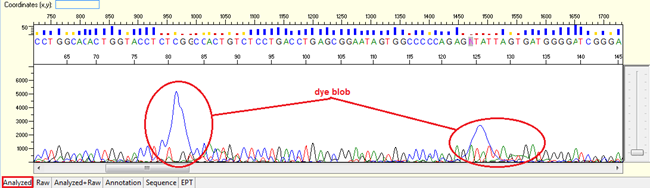
Causes:
-
- Unincorporated dye terminator molecules that did not remove efficiently during the steps of post sequencing clean-up.
- Degraded DNA template.
Solutions:
-
- When we see dye blobs, we will automatically re-run the reaction. If you notice this in your results and you are very sure that the DNA template is freshly prepared, kindly contact us to request for the re-run.
- Use freshly prepare DNA template for re-sequencing.
Pattern: A clear double peak present at a specific position within the continuous read length (CRL).
Trace view for result with SNP using Sequence Scanner software:
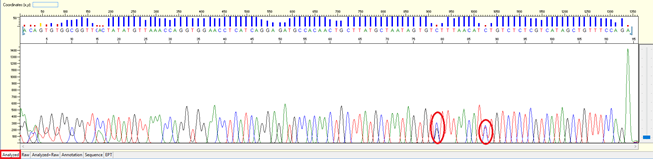
Cause:
-
- The nature of genomic DNA that carrying SNP in the different allele of the gene. When PCR, it generates mix of PCR products which have one base different.
Solution:
-
- SNP should not affect the analysis of a sequencing result. However, if one really wishes to get a clean sequence, DNA cloning will be an option before sequencing.
FAQ for DNA Barcoding Services
1st BASE Molecular Biology Services often coupled with data verification through Sanger DNA Sequencing. With appropriate control(s), molecular biology methods were employed to give more accurate and reproducible results and identifying the isolated biological samples down to the genus level. Other features of our DNA Barcoding Services are:
-
- Inclusive of sample preparation service with fast turnaround time
- Bi-directional sequencing prevents ambiguity in sequence
- Sequences are matched against a standard database, or a database of choice
- Comprehensive reports include sequences of sample, and the top 10 Hits of the BLAST results
- Neighbour Joining (unrooted tree)
- Additional analyses e.g. Phylogenetic analysis between samples is available as add-on services
Our DNA barcoding services are coupled with sample preparation services, allowing you to get the results in the shortest turnaround time, so that you can focus on the data analysis and decide the next move of your research experiments.
We accept isolated pure samples from bacteria, fungi, fish, insect, eukaryote, and plant.
Kindly please follow our sample preparation guidelines for DNA Barcoding Service to submit your samples after the order placed online.
If your sample type is not listed within our sample preparation guidelines, kindly contact us directly for more information.
We offer custom DNA Barcoding Services for any specific gene of your interest. Minimum of 5 samples for each specific gene are required for such customization.
Results will be downloaded through your Online Ordering account. The compressed file in .zip format (for Windows), contains the results with (i) DNA Barcoding Report, (ii) Assembled DNA Sequence(s), (iii) Top 10 Hit BLAST Results, and (iv) Unroot Phylogenetic Tree.
For more FAQs related to DNA Barcoding Services, please refer to this link FAQ for DNA Barcoding Services
FAQs on Avian DNA Testing

Please read our sample preparation guidelines for Avian DNA Testing before place the order online.
There are three different local language available to ease your understanding.
Our Avian DNA Sexing Test is designed to determine the gender from all kinds of non-ratite birds. Parrot is non-ratite birds and thus its gender can be determined through our designed molecular based DNA testing services.
The Avian DNA Test Report will be downloaded through your Online Ordering account. It contains the result interpretation and printable version of Bird DNA Gender Card(s). No hard copy results will be delivered.
For more FAQs related to Avian DNA Testing, please refer to this link FAQs on Avian DNA Testing
CONTACT US
Taman Perindustrian Bukit Serdang,
43300 Seri Kembangan,
Selangor, Malaysia
Copyright to © 2022 Apical Scientific Sdn. Bhd.